What is a heteroduplex?
A heteroduplex is a double stranded sequence comprised of two non-complementary strands. During the annealing step of PCR, non-complementary, but highly similar, DNA strands can form a heteroduplex. In other words, heteroduplexes are a byproduct of amplifying different templates in the same reaction.
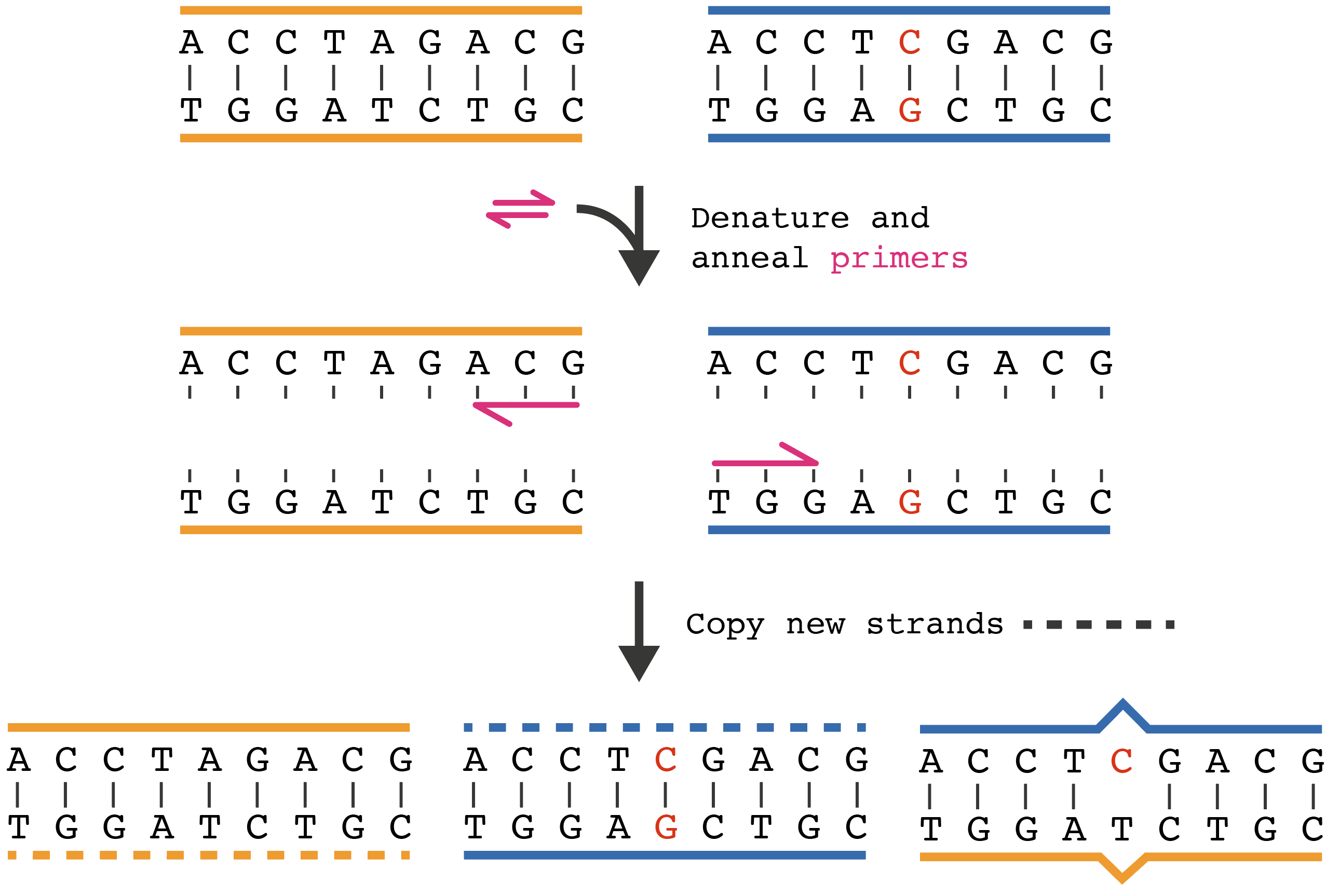
A heteroduplex is not a PCR chimera, which is defined on Wikipedia as
It occurs when the extension of an amplicon is aborted, and the aborted product functions as a primer in the next PCR cycle. The aborted product anneals to the wrong template and continues to extend, thereby synthesizing a single sequence sourced from two different templates.
What is heteroduplex splitting?
Starting with ccs v6.3.0, --hd-finder activates algorithms to detect heteroduplexes during the HiFi generation. Substitutions and large insertions (>20bp) with a significant strand bias are detected at the subread level. Subreads are aligned to the draft, and a pileup is generated. Divergent substitution sites are identified, and fisher’s exact test is used to determine if a substitution has strand bias. ZMWs labeled as heteroduplex are split, on-the-fly, into single-stranded CCS reads. As a consequence, ccs distinguish between double-stranded (DS) and single-stranded (DS) ZMWs and their consensus reads. Implications:
- Heteroduplex splitting is non-reversible
- The BAM output file will have three read groups instead of one
- Summary logs report double-strand and single-strand metrics
ccs_reports.txtfile contains two columns, double-strand and single-strand reads--by-strandand–hd-finderare non-equivalent, results can differ for the same ZMW
Additional read groups in BAM
The BAM file contains two different kinds of reads, single-strand and double-strand reads. Single-strand reads follow the by-strand scheme with /fwd and /rev name suffixes and ccs generates up to two single-strand reads per ZMW. Double-strand reads have no special distinguishing factor. Each of the three types of stranded reads have their own read groups. Single-stranded reads have an additional field in the DS tag of the read group. Simplified example
@RG ID:793f140b PL:PACBIO DS:READTYPE=CCS;STRAND=FORWARD <- single-strand reads /fwd
@RG ID:36fc54d5 PL:PACBIO DS:READTYPE=CCS;STRAND=REVERSE <- single-strand reads /rev
@RG ID:5d30364d PL:PACBIO DS:READTYPE=CCS <- double-strand reads
Summary logs
At the end of each execution, ccs reports for --log-level INFO a summary. This summary contains combined and individual metrics for DS and SS.
-------------------------------------------------
Summary stats abbreviations:
ZMW - A productive Zero-Mode Waveguide
DS - Double Strand
SS - Single Strand
DS-ZMW - All subreads were used from a single ZMW
SS-ZMW - ZMW is split into fwd and rev strands,
each strand is polished individually
DS-Read - CCS read of a DS-ZMW
SS-Read - CCS read of one strand of a SS-ZMW
HiFi - CCS reads with predicted accuracy >=Q20
UMY - Unique Molecular Yield of all reads passing filters
HiFi Yield - UMY of >=Q20 DS- and SS-ZMWs, longest read per ZMW
-------------------------------------------------
ZMWs Input : 53895
ZMWs Written : 22684
- DS / SS : 22644 / 40
UMY : 413.2 MBases (6.8 GBases/hr)
- DS / SS : 412.4 MBases / 733.7 KBases
HiFi Yield : 413.5 MBases (6.8 GBases/hr)
- DS / SS : 412.4 MBases / 1.0 MBases
HiFi Reads : 22701
- DS / SS : 22644 / 57
HiFi Avg Size : 18.2 KBases
HiFi Avg QV : 30.2
Strand-aware ccs_reports.txt
Typical content of the strand-aware ccs_reports.txt file. Contrary to the default output, this file does not report numbers in ZMWs, but actual DS and SS reads. Accounting in SS ZMWs is not possible, as one strand might fail and the other succeed. The percentage of the Inputs is with respect to the number of ZMWs, all other percentages are with respect to reads in their column.
Double-Strand Reads Single-Strand Reads
Inputs : 53590 (99.43%) 609 (0.564%)
Passed : 22644 (42.25%) 57 (9.360%)
Failed : 30946 (57.75%) 552 (90.64%)
Tandem repeats : 461 (1.490%) 0 (0.000%)
Exclusive failed counts
Below SNR threshold : 870 (2.811%) 0 (0.000%)
Median length filter : 0 (0.000%) 0 (0.000%)
Shortcut filters : 0 (0.000%) 0 (0.000%)
Lacking full passes : 26226 (84.75%) 0 (0.000%)
Coverage drops : 30 (0.097%) 0 (0.000%)
Insufficient draft cov : 61 (0.197%) 310 (56.16%)
Draft too different : 0 (0.000%) 0 (0.000%)
Draft generation error : 173 (0.559%) 54 (9.783%)
Draft above --max-length : 0 (0.000%) 0 (0.000%)
Draft below --min-length : 0 (0.000%) 0 (0.000%)
Reads failed polishing : 0 (0.000%) 0 (0.000%)
Empty coverage windows : 3 (0.010%) 0 (0.000%)
CCS did not converge : 2 (0.006%) 0 (0.000%)
CCS below minimum RQ : 3581 (11.57%) 188 (34.06%)
Unknown error : 0 (0.000%) 0 (0.000%)
Can I combine it with HiFi kinetics?
Yes! Check out kinetics FAQ