30,000-foot overview
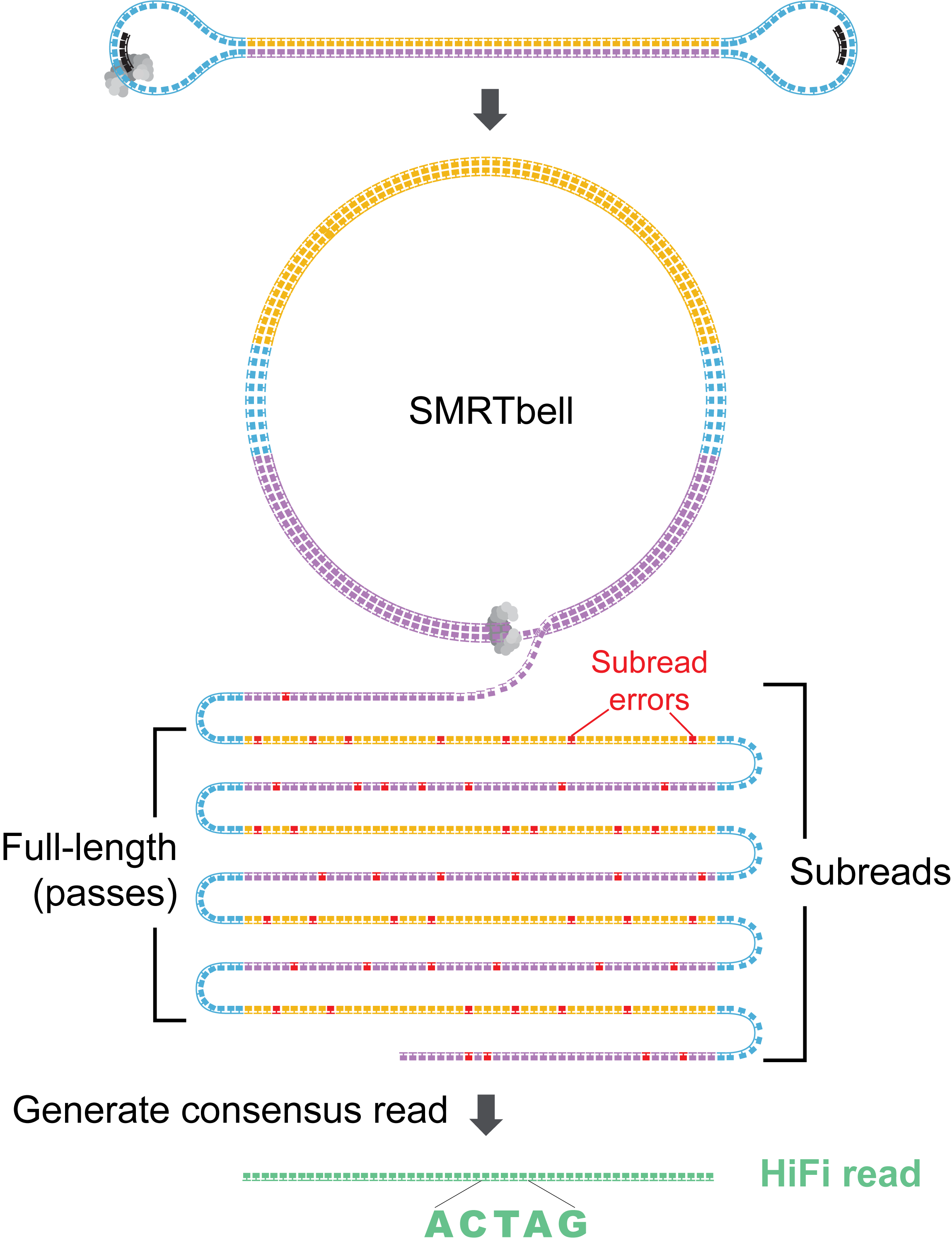
How does ccs generate HiFi reads?
Each SMRTbell molecule is processed in two main stages:
- Generate an approximate draft consensus from a few subreads to provide a starting template for polishing.
- Iteratively polish the template using all subreads to generate a final consensus sequence.
The more detailed steps for a single ZMW are as follows.
1. Initial filtering
Filter ZMWs:
- Remove ZMWs with signal-to-noise ratio (SNR) below
--min-snr.
Filter subreads:
- Remove subreads with lengths <50% or >200% of the median subread length.
- Stop if number of full-length subreads is fewer than
--min-passes.
Why does the subread length filter use 50% and 200% thresholds?
Subreads that are longer than 200% of the median length are likely due to missed adapter calls, where the initial polymerase read was wrongly split into subreads. Subreads shorter than 50% (“partial” passes) are a challenge to map confidently against the template, especially when the template is a palindromic sequence. To avoid improper mappings, short subreads are excluded.
2. Draft generation
The polish stage iteratively improves upon a candidate template sequence. Because polishing is very compute intensive, it is desirable to start with a template that is as close as possible to the true sequence of the molecule to reduce the number of iterations until convergence. The ccs software does not pick a full-length subread as the initial template to be polished, but instead generates an approximate draft consensus sequence using our improved implementation of the Sparc graph consensus algorithm. This algorithm depends on a subread to backbone alignment that is generated by our own mapper pancake using edlib as the core aligner. Typically, subreads have accuracy of around 90% and the draft consensus has a higher accuracy, but is still below 99%.
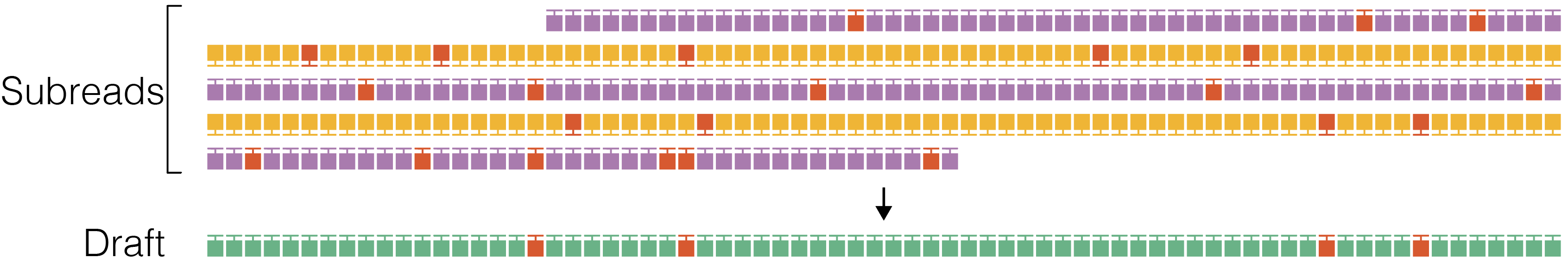
Stop if draft length is shorter than --min-length and longer than --max-length.
3. Alignment
Align subreads to the draft consensus using pancake with KSW2 for downstream windowing and filtering.
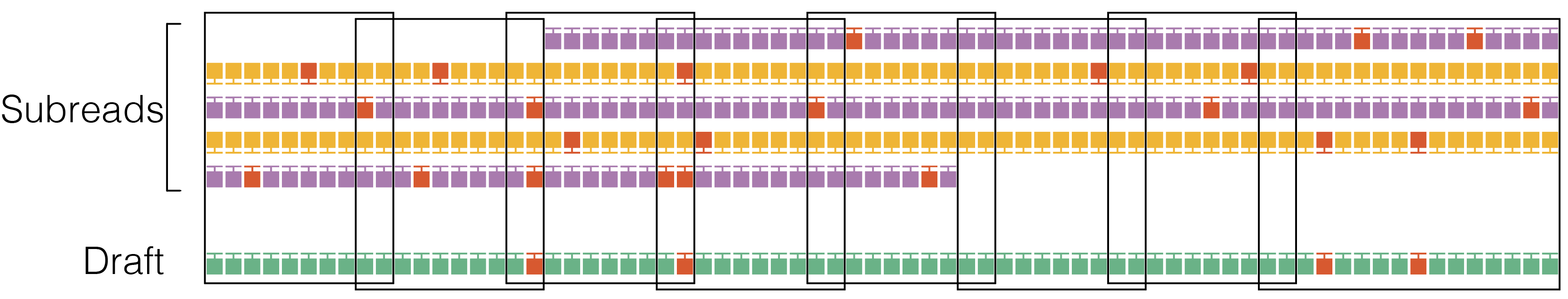
4. Windowing
Divide the the subread-to-draft alignment into overlapping windows with a target size of 22 bp with ±2 bp overlap. Avoid breaking windows at simple repeats (homopolymers to 4-mer repeats) to reduce edge cases at window borders. Windowing reduces the algorithm run time from quadratic to linear in the insert size.
5. Single-strand artifacts
Identify heteroduplexes, where one strand of the SMRTbell differs significantly from the reverse complement of the other strand. Subread orientation is inferred from the alignment. Small heteroduplexes, e.g., single base A paired with a matching G, are retained and the ambiguity is reflected in base quality. Molecules with a single difference longer than 20 bp between the strands are removed and recorded as heteroduplexes in the <outputPrefix>.ccs_report.txt file.
6. Trim large insertions
Insertions in the subreads relative to the draft that are longer than --max-insertion-size, default 30 bp, are trimmed since they typically represented spurious sequencing activity and confound window-based, iterative polishing.
7. Candidate filtering
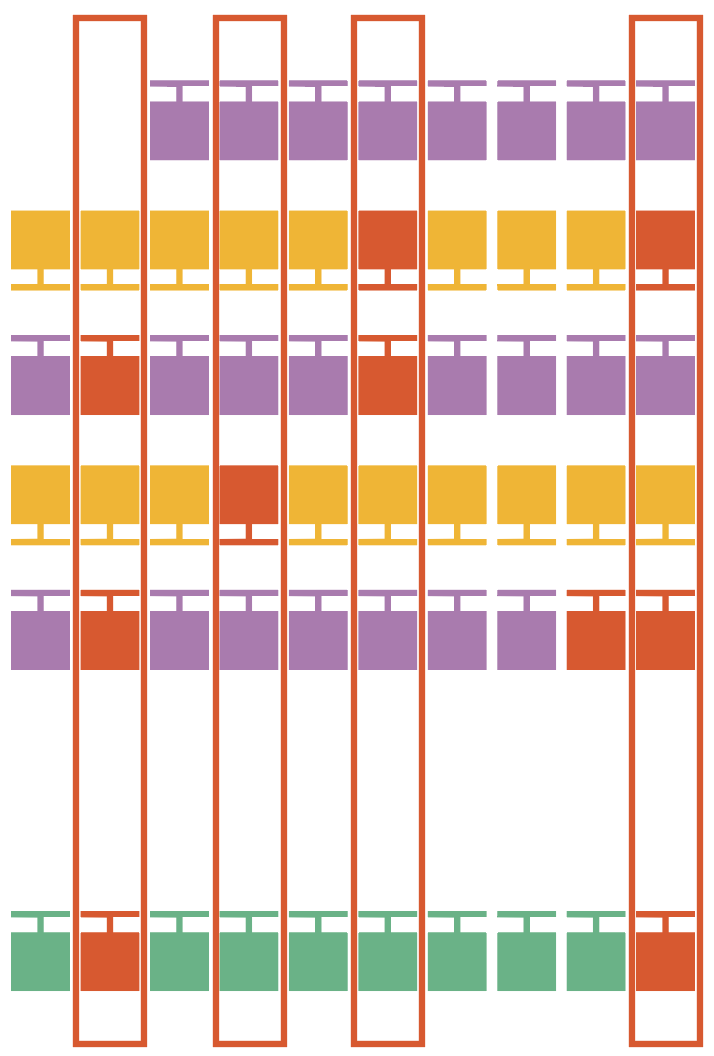
8. Polishing
The core polishing uses the arrow algorithm, a left-right Hidden Markov-Model (HMM) that models the enzymatic and photophysical events in SMRT sequencing. Emission and transition parameters are estimated by a dinucleotide template context. Transition parameters form a homogeneous Markov chain. In other words, the transition parameters do not depend on the position within the template, only the pulse width of a base call, the dinucleotide context of the template, and the SNR of the ZMW. Arrow computes the log-likelihood that the subread originates from the template, marginalizing over all possible alignments of the subread to the template. For every position in the template that is a candidate for polishing, arrow tests if the log-likelihood is improved by substituting one of the other three nucleotides, inserting one of the four nucleotides after the position, or deleting the position itself. Once arrow does not find any further beneficial mutations to the template in an iteration, it stops.
9. QV calculation
The log-likelihood ratio between the most likely template sequence and all of its mutated counterparts is used to calculate a quality for each base in the final consensus. The average of the per-base qualities is the read accuracy, rq.
10. Final
To finalize the output, the per-window consensus template sequences and base qualities are concatenated and overhangs, overlaps between adjacent windows, are trimmed. If the predicted read accuracy is at least --min-rq then the consensus read is written to the output.