How do I read the ccs_report.txt file?
By default, each CCS run generates a <outputPrefix>.ccs_report.txt file. This file summarizes how many ZMWs generated HiFi reads (CCS) and how many failed CCS generation because of the listed causes. For those failing, each ZMW contributes to exactly one reason of failure; percentages are with respect to number of failed ZMWs.
The following comments refer to the filters that are explained in the FAQ above.
ZMWs input : 1671
ZMWs pass filters : 824 (49.31%) ┐
ZMWs fail filters : 104 (6.22%) | Sum to 100%
ZMWs shortcut filters : 743 (44.46%) <- Low-pass ZMWs skipped by --all ┘
ZMWs with tandem repeats : 10 (0.60%) <- With repeats larger than --min-tandem-repeat-length
Exclusive failed counts
Below SNR threshold : 30 (28.85%) <- SNR below --min-snr.
Median length filter : 0 (0.00%) <- All subreads are <50% or >200% of the median subread length
Lacking full passes : 0 (0.00%) <- Fewer than --min-passes full-length (FL) reads
Heteroduplex insertions : 10 (9.62%) <- Single-strand artifacts
Coverage drops : 43 (41.35%) <- Coverage drops would lead to unreliable polishing results
Insufficient draft cov : 16 (15.38%) <- Not enough subreads aligned to draft end-to-end
Draft too different : 0 (0.00%) <- Fewer than --min-passes FL reads aligned to draft
Draft generation error : 3 (2.88%) <- Subreads don't agree to generate a draft sequence
Draft above --max-length : 0 (0.00%) <- Draft sequence is longer than --max-length
Draft below --min-length : 0 (0.00%) <- Draft sequence is shorter than --min-length
Reads failed polishing : 0 (0.00%) <- Too many subreads were dropped while polishing
Empty coverage windows : 2 (1.92%) <- At least one window has no coverage
CCS did not converge : 0 (0.00%) <- Draft has too many errors that can't be polished in time
CCS adapter concatenation : 0 (0.000) <- Reads which are a concatenation of the adapter
CCS adapter palindrome : 0 (0.000) <- Miscalled adapters with reverse complemented inserts
CCS adapter residue : 0 (0.000) <- Reads that have one or more adapters close to either end
ZMW with full-length subread : 0 (0.000) <- fail_reads.bam reads that didn't generate consensus reads
ZMW with control failure : 0 (0.000) <- Reads that failed polishing and are spike-in controls
ZMW with control success : 0 (0.000) <- Reads that passed polishing and are spike-in controls
CCS below minimum RQ : 0 (0.00%) <- Predicted accuracy is below --min-rq
Unknown error : 0 (0.00%) <- Rare implementation errors
Additional passing metrics
ZMWs missing adapters : 0 (0.000%)
- - - - - - - - - - - - - - - : - - - - -
HiFi Reads : 6
HiFi Yield (bp) : 63,881
HiFi Read Length (mean, bp) : 10,646
HiFi Read Length (median, bp) : 10,289
HiFi Read Length N50 (bp) : 10,439
HiFi Read Quality (median) : 31
HiFi Number of Passes (mean) : 9
<Q20 Reads : 2
<Q20 Yield (bp) : 23,342
<Q20 Read Length (mean, bp) : 11,671
<Q20 Read Length (median, bp) : 11,671
<Q20 Read Quality (median) : 18
>=Q30 Reads : 5
>=Q30 Yield (bp) : 54,336
>=Q30 Read Length (mean, bp) : 10,867
>=Q30 Read Length (median, bp): 10,439
>=Q30 Read Quality (median) : 31
Base quality >=Q30 (bp) : 62,526 (97.9%)
If run in --by-strand mode, please have a look at the by-strand FAQ.
If run in --split-heteroduplexes mode, please have a look at the strand-aware FAQ.
Coverage drops
Example for a coverage drop in a single ZMW, subreads colored by strand orientation:
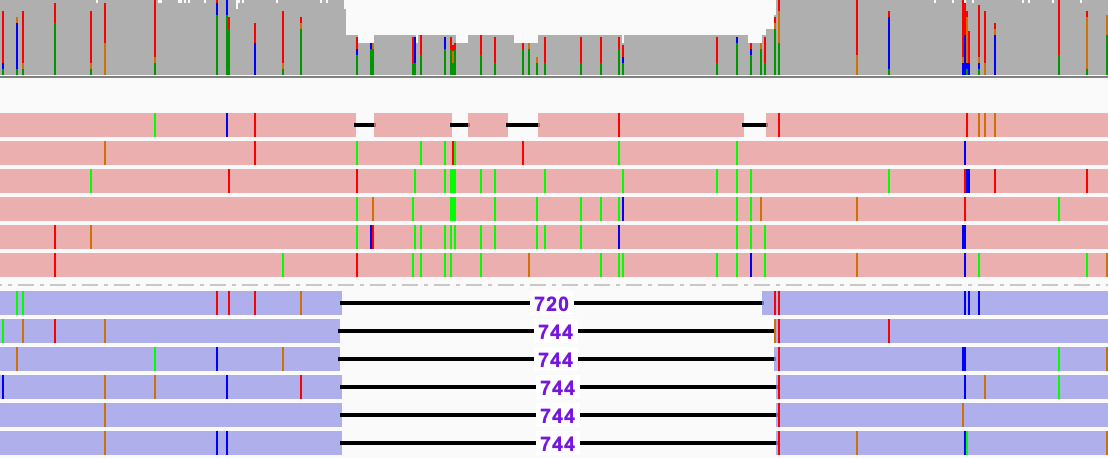
During sequencing of the molecule, one strand exhibits 744 more bases than its reverse complemented strand. What happened? Either there is a gain or loss of information. An explanation for loss of information could be that a secondary structure, the 744 bp forming a hairpin, could affect the replication during PCR and lead to loss of bases. Gain of information could also happen during PCR, when the polymerase gets stuck and incorporates the current base too often. In this example, there is a homopolymer of 744 A bases. While it might be obvious to a human eye what happened, its not the responsibility of ccs to interpret and recover molecular damage. Even if there were a low-complexity filter for those regions, setting the appropriate threshold would be arbitrary; would a 10bp homopolymer insertion be valid, but 11bp would get discarded?
How do I read the zmw_metrics.json file?
Per default, each ccs run generates a <outputPrefix>.zmw_metrics.json.gz file. Change file name with --metrics-json. The resulting *.zmw_metrics.json file has one field zmws that contains an array. Each entry in this array describes one input ZMW. You can pretty print its content with
zcat out.zmw_metrics.json.gz | python3 -m json.tool
Example content with two ZMWs:
{
"zmws": [
{
"effective_coverage": 8.2,
"has_tandem_repeat": false,
"insert_size": 12972,
"num_full_passes": 7,
"polymerase_length": 111325,
"predicted_accuracy": 0.998,
"status": "SUCCESS",
"wall_end": 5529991,
"wall_start": 91,
"zmw": "m64011_200315_001815/0"
},
{
"effective_coverage": 1,
"has_tandem_repeat": true,
"insert_size": 12783,
"num_full_passes": 1,
"polymerase_length": 27511,
"predicted_accuracy": -1.0,
"status": "TOO_FEW_PASSES",
"wall_end": 2425263,
"wall_start": 1179648,
"zmw": "m64011_200315_001815/2"
},
]
}
Each ZMW consists of following fields:
status, did CCS finish withSUCCESSor fail with an error (identical order as inccs_reports.txt):POOR_SNR, All subreads were below SNR threshold--min-snr(1)NO_SUBREADS, All subreads were filtered by the median length filter (2)TOO_FEW_PASSES, Fewer than--min-passesfull-length (FL) reads (3)LOW_PASS_SHORTCUT, ZNW skipped polishing, active with--allHETERODUPLEXES, Single-strand artifacts, see definition of a heteroduplexCOVERAGE_DROPS, Coverage drops against draft would lead to unreliable polishing resultsINSUFFICIENT_SPANS, Not enough subreads aligned to draft end-to-endTOO_FEW_PASSES_AFTER_DRAFT_ALIGNMENT, Fewer than--min-passesFL reads aligned to draftDRAFT_FAILURE, Subreads don’t agree to generate a draft sequenceTOO_LONG, Draft sequence is longer than--max-length(4)TOO_SHORT, Draft sequence is shorter than--min-length(4)TOO_MANY_UNUSABLE, Too many subreads were dropped while polishingEMPTY_WINDOW_DURING_POLISHING, At least one window has no coverageNON_CONVERGENT, Draft has too many errors that can’t be polished in timePOOR_QUALITY, Predicted accuracy is below –min-rq (5)EXCEPTION_THROWN, Rare implementation errors
effective_coverage, average coverage used for polishing, more info herehas_tandem_repeat, sDUST found a tandem repeat larger than--min-tandem-repeat-length Ninsert_size, the length – in precedence order – of- the polished sequence if polishing was successful or
- the draft sequence if draft generation was successful or
- the subread of median length
num_full_passes, number of full-length subreads used, more info herepolymerase_length, total number of bases produced from a ZMW after trimming the low-quality regions, including adapterspredicted_accuracy, also known as read quality, set to-1if not calculatedwall_end, start of last base of the polymerase read in approximate raw frame count since start of moviewall_start, start of first base of the polymerase read in approximate raw frame count since start of moviezmw, the ZMW ID
Additional fields starting with _ are implementation details and are subject to change.
Is there a progress report?
Yes. With --log-level INFO, ccs provides status to stderr every --refresh-rate seconds (default 5):
37440/14155/55.3 20975/7960/31.1 2h 27m
The log output explains each field:
Logging info: Z1/Z2/Z3 C1/C2/C3 ETA
Z1: #ZMWs processed since start
Z2: #ZMWs processed in the last minute
Z3: #ZMWs processed in the last minute per thread
C1: #CCSs generated since start
C2: #CCSs generated in the last minute
C3: #CCSs generated in the last minute per thread
ETA: Estimated remaining run time, ~extrapolated
If there is no .pbi file present, ETA will be omitted.